
Mitochondrial Transplantation Modulates Inflammation and Apoptosis, Alleviating Tendinopathy Both In Vivo and In Vitro

 , and
, and 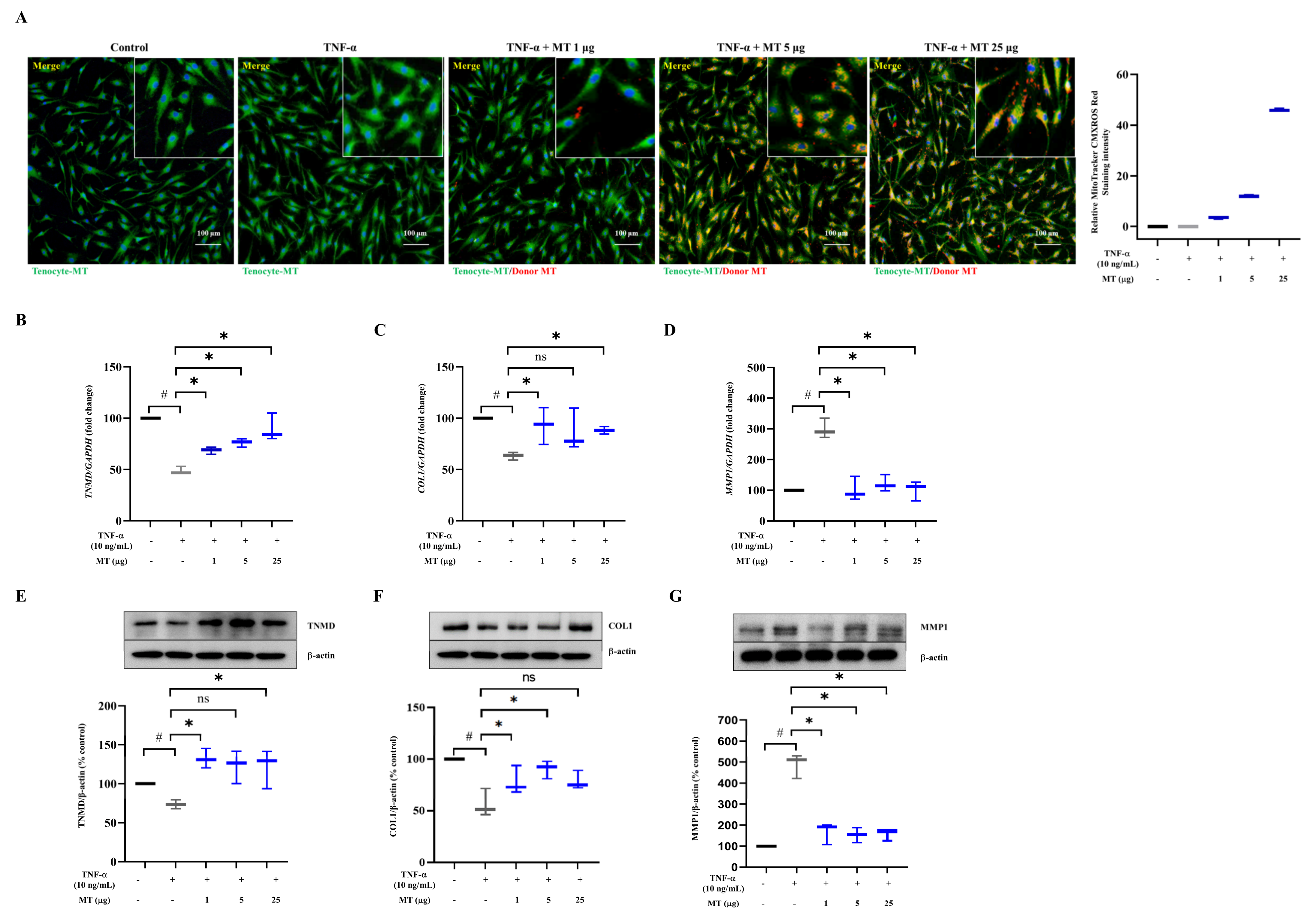{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Establishment of a Damaged Tenocyte Model
2.3. Isolation of Mitochondria
2.4. Transfer of Isolated Mitochondria on Tenocytes In Vitro
2.5. Verification of the Incorporation of Exogenous Mitochondria into Tenocytes
2.6. Establishment of an In Vivo Tendinopathy Model
2.7. Injection of Isolated Mitochondria In Vivo
2.8. Confirmation of Mitochondrial Transplantation In Vivo
2.9. ROS Measurement
2.10. Mitochondrial ROS (mROS) Measurement
2.11. Adenosine Triphosphate (ATP) Measurement
2.12. Mitochondrial Membrane Potential
2.13. Real-Time Polymerase Chain Reaction (PCR) Analysis
2.14. Western Blot Analysis
2.15. Cytokine Assay
2.16. Microarray Analysis
2.17. Hydroxyproline Assay
2.18. Mdivi-1 Treatment
2.19. Statistical Analysis
2.20. Data Availability
3. Results
3.1. Successful Mitochondrial Isolation and Transplantation into Tenocytes Confirmed by Expression of Mitochondrial Markers In Vitro
3.2. Mitochondrial Transplantation Enhanced TNMD and COL1 Expression and Decreased MMP1 Expression in TNFα-Treated Tenocytes In Vitro
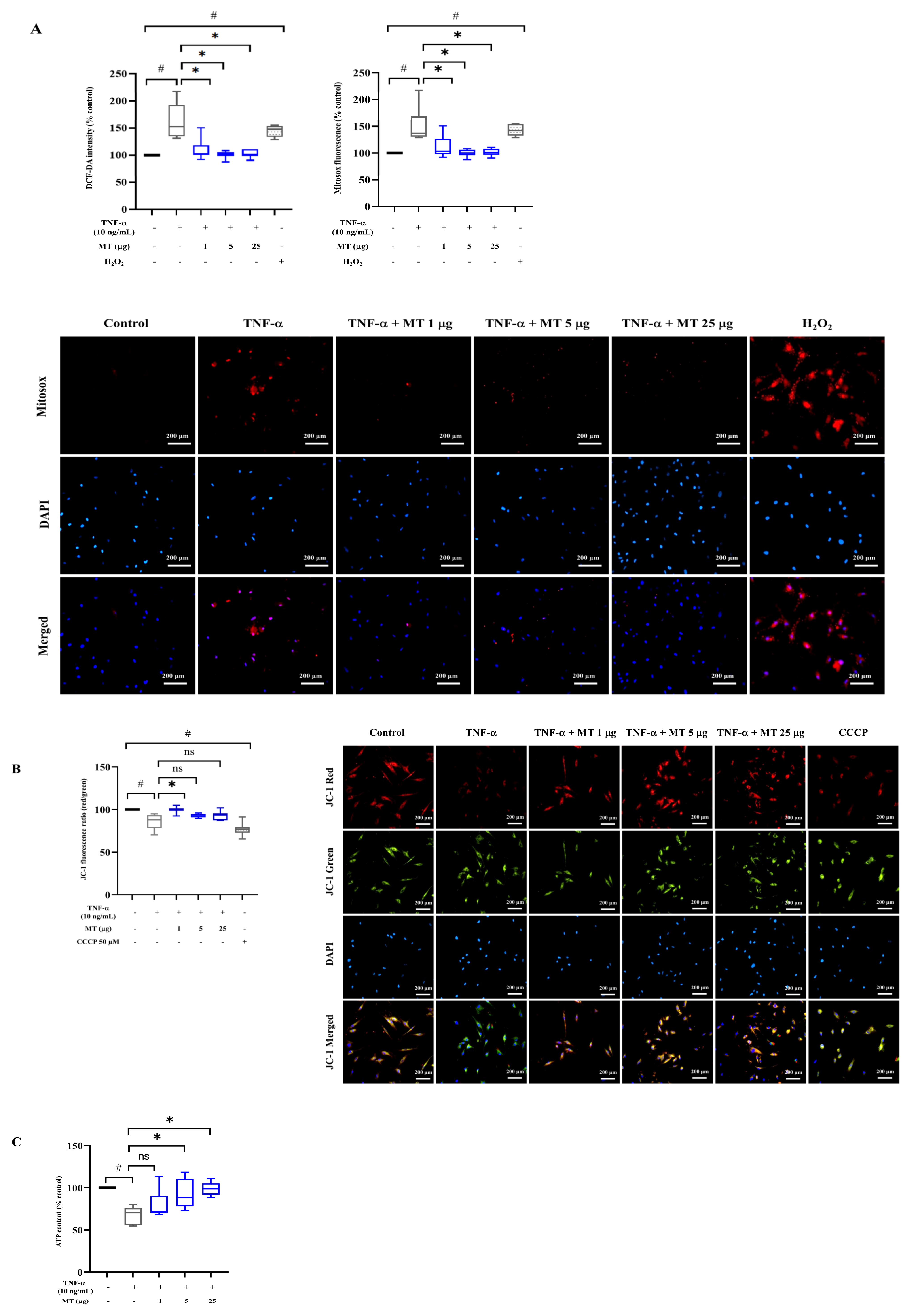
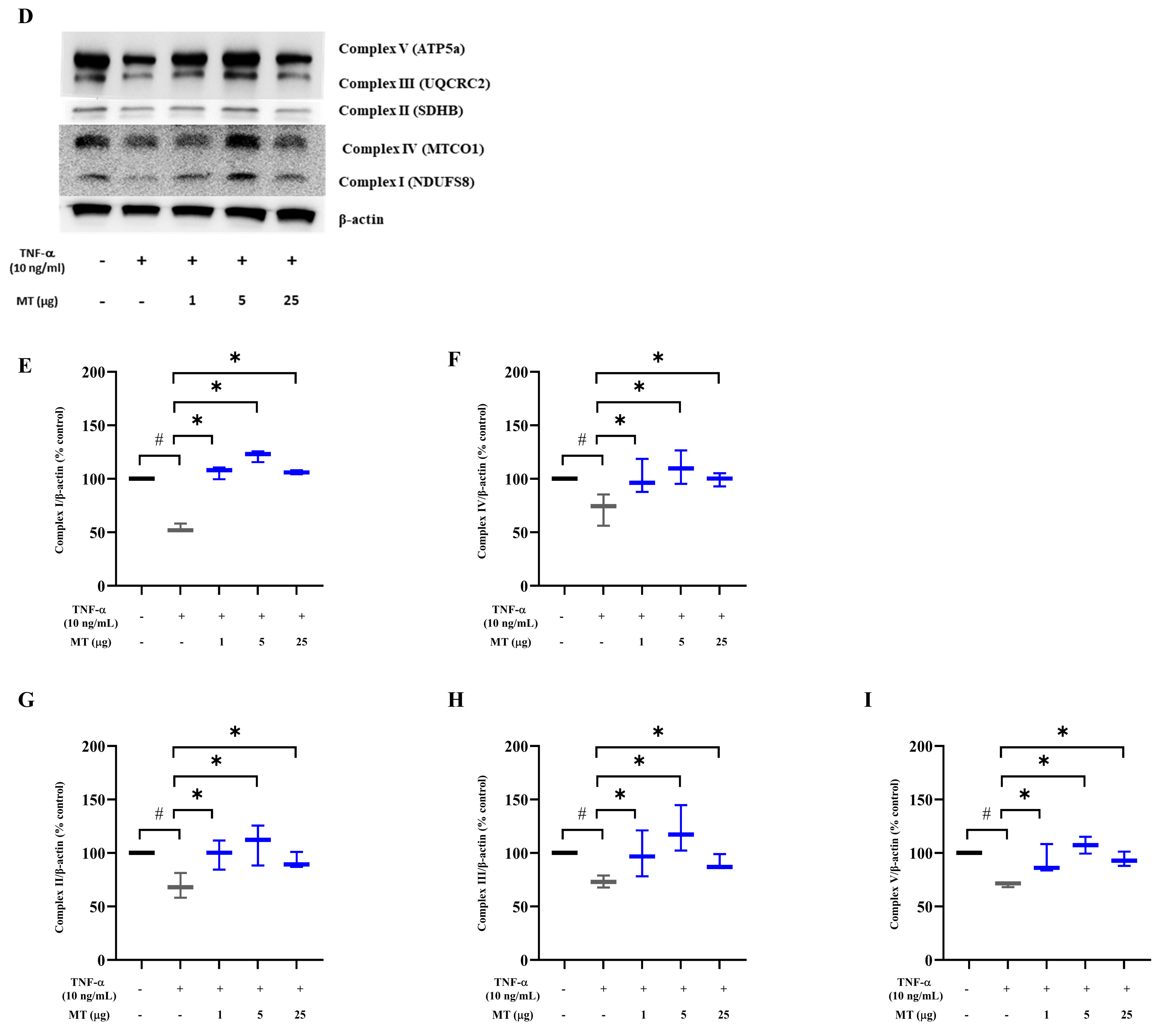
3.3. Mitochondrial Transplantation Attenuated Intracellular Oxidative Stress in TNF-α-Treated Tenocytes In Vitro
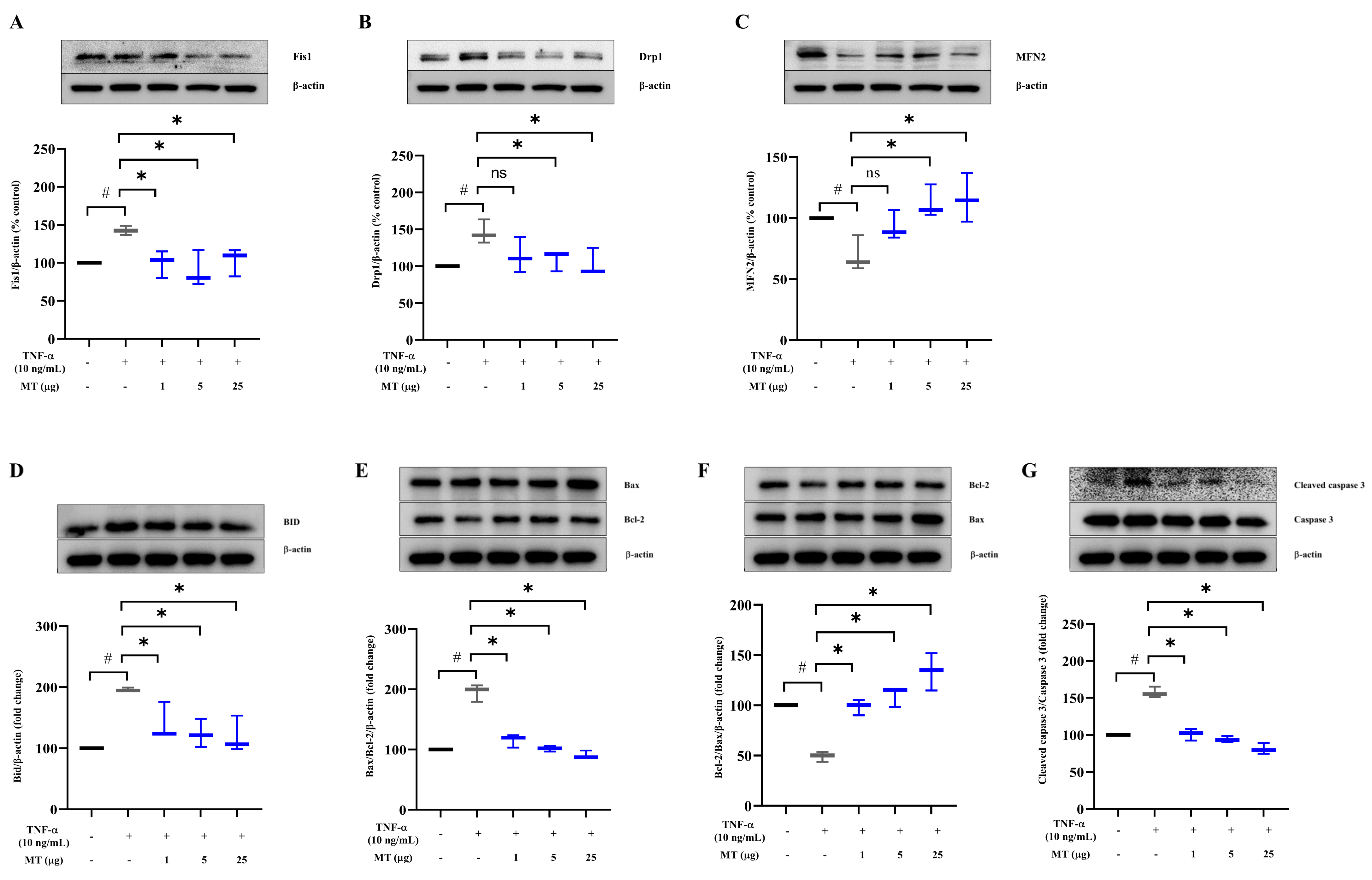
3.4. Mitochondrial Transplantation Downregulated Fission Factors and Upregulated Fusion Factors in TNFα-Treated Tenocytes In Vitro
3.5. Mitochondrial Transplantation Inhibited Apoptosis in TNFα-Treated Tenocytes In Vitro
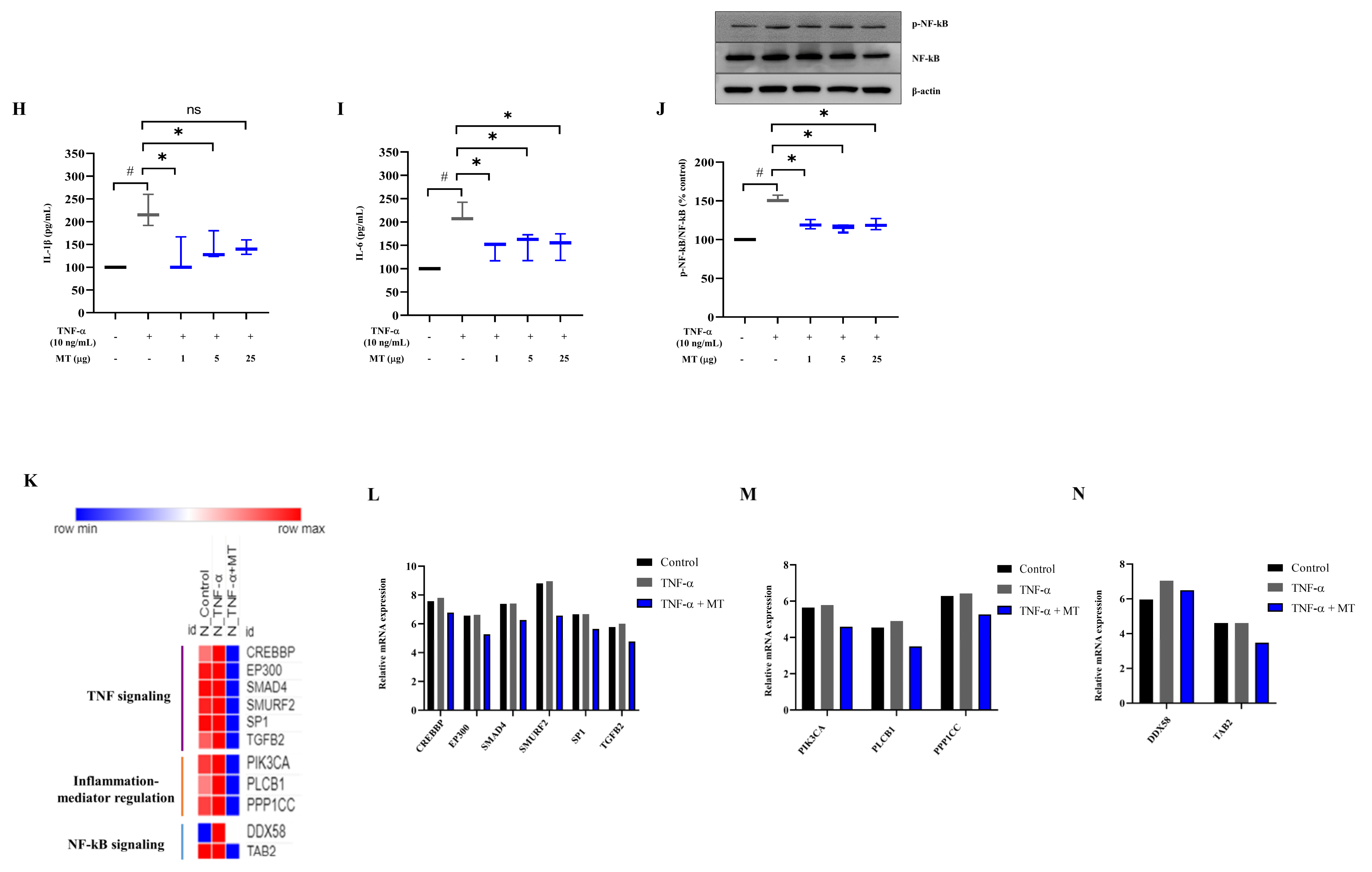
3.6. Mitochondrial Transplantation Inhibited Inflammatory Marker Expression in TNFα-Treated Tenocytes In Vitro
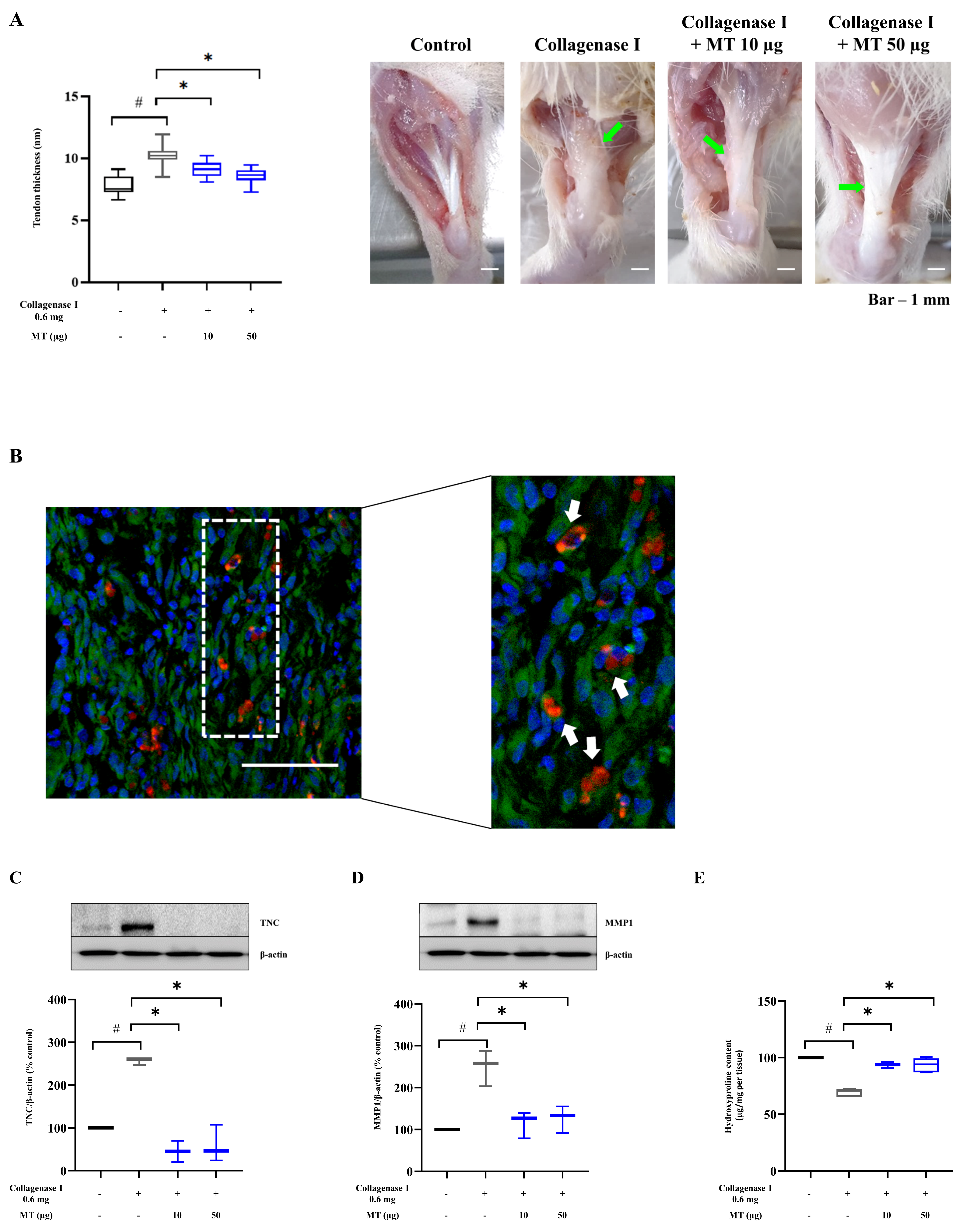
3.7. Mitochondrial Transplantation Alleviated Collagenase-Induced Tendinopathy in Rats In Vivo
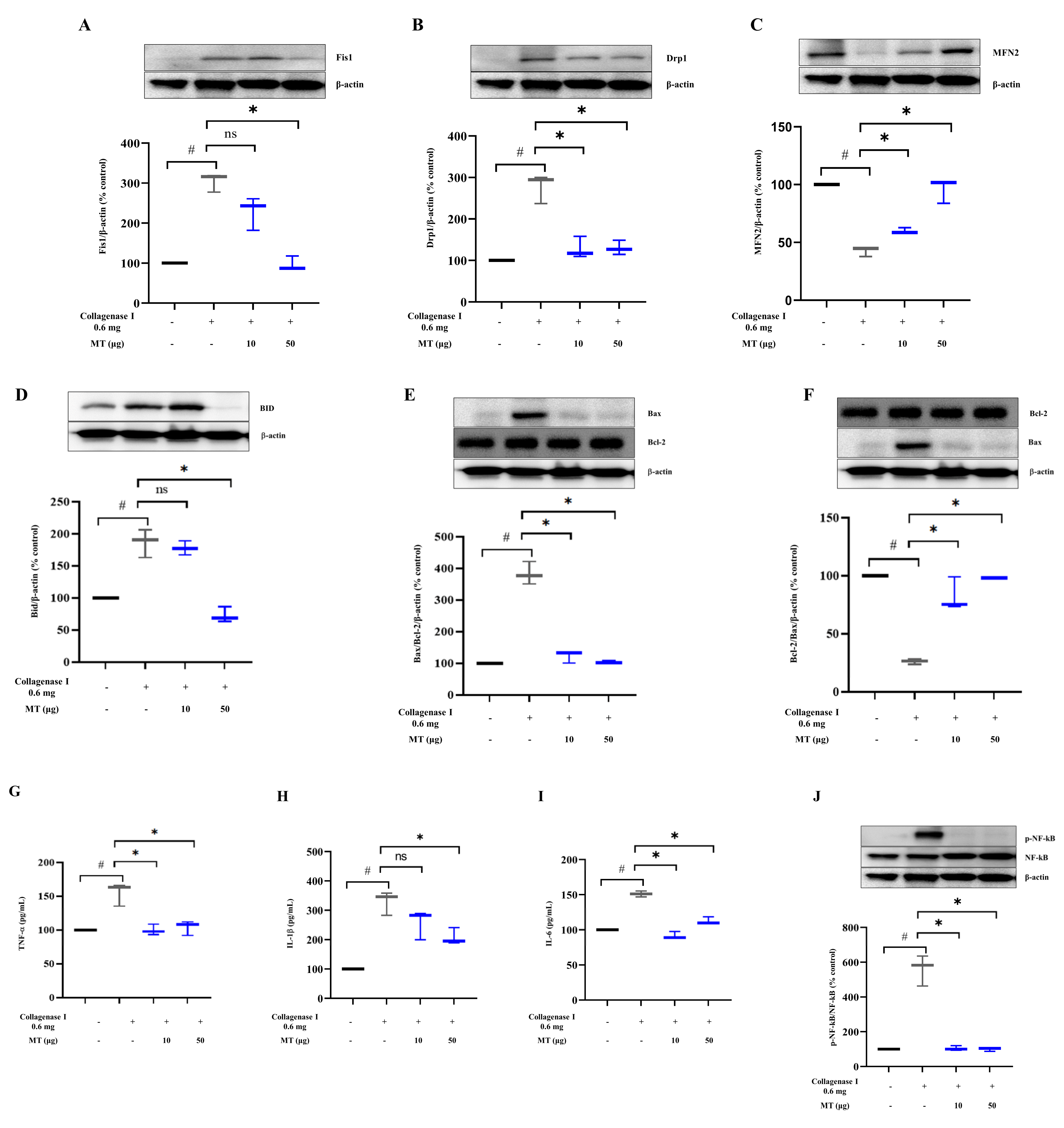
3.8. Mitochondrial Transplantation Promoted Mitochondrial Dynamics and Inhibited Apoptosis and Inflammation in Collagenase-Induced Tendinopathy in Rats In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATP | adenosine triphosphate |
| ATP5a | ATP synthase F1 subunit alpha |
| Bax | Bcl-2 associated X |
| Bcl-2 | B-cell lymphoma 2 |
| BID | BH3-interacting domain death agonist |
| BSA | bovine serum albumin |
| CCCP | carbonyl cyanide m-chlorophenyl hydrazone |
| COL1 | collagen 1 |
| DAPI | 4′6-diamidino-2-phenylindole |
| DMAB | dimethylamino benzaldehyde |
| DCF-DA | dichlorodihydrofluorescein diacetate |
| DPBS | Dulbecco’s phosphate-buffered saline |
| DRP1 | dynamin-related protein 1 |
| EGTA | ethylene glycol tetra acetic acid |
| ELISA | enzyme-linked immunosorbent assay |
| Fis1 | fission 1 |
| H2O2 | hydrogen peroxide |
| HCl | hydrogen chloride |
| HEPES | hydroxyethyl piperazine ethanesulfonic acid |
| HRP | horseradish peroxidase |
| IACUC | Institutional Animal Care and Use Committee |
| IL-1β | interleukin 1 beta |
| IL-6 | interleukin 6 |
| MFN2 | mitofusin 2 |
| MitoTracker CMXRos red | CMXRos red |
| MMP | mitochondrial membrane potential |
| MMP1 | matrix metalloproteinase-1 |
| MT | mitochondria |
| MTCO1 | mitochondrially encoded cytochrome C oxidase 1 |
| NaOH | sodium hydroxide |
| NDUFS8 | NADH ubiquinone oxidoreductase core subunit S8 |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| OXPHOS | oxidative phosphorylation |
| P/S | penicillin/streptomycin |
| PBS | phosphate-buffered saline |
| Polyacrylamide gel electrophoresis | PAGE |
| PVDF | polyvinylidene difluoride |
| RIPA | radio immunoprecipitation assay |
| ROS | reactive oxygen species |
| SDHB | succinate dehydrogenase complex iron-sulfur subunit B |
| SDS | sodium dodecyl sulfate |
| TMB | tetramethylbenzidine |
| TNC | tenascin C |
| TNF-α | tumor necrosis factor-α |
| TNMD | tenomodulin |
| UC-MSC | Umbilical cord-mesenchymal stem cell |
| UQCRC2 | ubiquinol-cytochrome c reductase core protein 2 |
References
- Maffulli, N.; Wong, J.; Almekinders, L.C. Types and epidemiology of tendinopathy. Clin. Sports Med. 2003, 22, 675–692. [Google Scholar] [CrossRef]
- Dimitrios, S. Exercise for tendinopathy. World J. Methodol. 2015, 5, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Abat, F.; Alfredson, H.; Cucchiarini, M.; Madry, H.; Marmotti, A.; Mouton, C.; Oliveira, J.M.; Pereira, H.; Peretti, G.M.; Romero-Rodriguez, D.; et al. Current trends in tendinopathy: Consensus of the ESSKA basic science committee. Part I: Biology, biomechanics, anatomy and an exercise-based approach. J. Exp. Orthop. 2017, 4, 1–11. [Google Scholar] [CrossRef]
- Andres, B.M.; Murrell, G.A. Treatment of tendinopathy: What works, what does not, and what is on the horizon. Clin. Orthop. Relat. Res. 2008, 466, 1539–1554. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Maffulli, N. Tendon injury and tendinopathy: Healing and repair. J. Bone Jt. Surg. 2005, 87, 187–202. [Google Scholar] [CrossRef] [Green Version]
- Weber, S.; Chahal, J. Management of Rotator Cuff Injuries. J. Am. Acad. Orthop. Surg. 2020, 28, e193–e201. [Google Scholar] [CrossRef]
- Abate, M.; Silbernagel, K.G.; Siljeholm, C.; Di Iorio, A.; De Amicis, D.; Salini, V.; Werner, S.; Paganelli, R. Pathogenesis of tendinopathies: Inflammation or degeneration? Arthritis Res. Ther. 2009, 11, 235. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Chen, Y.; Huang, J.; Zhao, K.; Chen, X.; Yin, Z.; Heng, B.C.; Chen, W.; Shen, W. The roles of inflammatory mediators and immunocytes in tendinopathy. J. Orthop. Transl. 2018, 14, 23–33. [Google Scholar] [CrossRef]
- Millar, N.L.; Murrell, G.A.; McInnes, I.B. Inflammatory mechanisms in tendinopathy–Towards translation. Nat. Rev. Rheumatol. 2017, 13, 110–122. [Google Scholar] [CrossRef]
- Karthikeyan, S.; Griffin, D.R.; Parsons, N.; Lawrence, T.M.; Modi, C.S.; Drew, S.J.; Smith, C.D. Microvascular blood flow in normal and pathologic rotator cuffs. J. Shoulder Elb. Surg. 2015, 24, 1954–1960. [Google Scholar] [CrossRef]
- Morais, D.S.; Torres, J.; Guedes, R.M.; Lopes, M.A. Current Approaches and Future Trends to Promote Tendon Repair. Ann. Biomed. Eng. 2015, 43, 2025–2035. [Google Scholar] [CrossRef]
- Benson, R.T.; McDonnell, S.M.; Knowles, H.J.; Rees, J.L.; Carr, A.J.; Hulley, P.A. Tendinopathy and tears of the rotator cuff are associated with hypoxia and apoptosis. J. Bone Jt. Sur. 2010, 92, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Fearon, U.; Canavan, M.; Biniecka, M.; Veale, D.J. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 385–397. [Google Scholar] [CrossRef]
- Millar, N.L.; Reilly, J.H.; Kerr, S.C.; Campbell, A.L.; Little, K.J.; Leach, W.J.; Rooney, B.P.; Murrell, G.A.; McInnes, I.B. Hypoxia: A critical regulator of early human tendinopathy. Ann. Rheum. Dis. 2012, 71, 302–310. [Google Scholar] [CrossRef]
- Lowes, D.A.; Wallace, C.; Murphy, M.P.; Webster, N.R.; Galley, H.F. The mitochondria targeted antioxidant MitoQ protects against fluoroquinolone-induced oxidative stress and mitochondrial membrane damage in human Achilles tendon cells. Free Radic. Res. 2009, 43, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Goda, N.; Kanai, M. Hypoxia-inducible factors and their roles in energy metabolism. Int. J. Hematol. 2012, 95, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Mabalirajan, U. Rejuvenating cellular respiration for optimizing respiratory function: Targeting mitochondria. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L103–L113. [Google Scholar] [CrossRef] [Green Version]
- Picard, M.; Wallace, D.C.; Burelle, Y. The rise of mitochondria in medicine. Mitochondrion 2016, 30, 105–116. [Google Scholar] [CrossRef]
- Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M.; Russell, O.M. Mitochondrial transplantation—A possible therapeutic for mitochondrial dysfunction? Mitochondrial transfer is a potential cure for many diseases but proof of efficacy and safety is still lacking. EMBO Rep. 2020, 21, e50964. [Google Scholar] [CrossRef]
- Yamada, Y.; Ito, M.; Arai, M.; Hibino, M.; Tsujioka, T.; Harashima, H. Challenges in Promoting Mitochondrial Transplantation Therapy. Int. J. Mol. Sci. 2020, 21, 6365. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Bruzzese, M.; Chou, S.H.; Ning, M.; Ji, X.; Lo, E.H. Extracellular Mitochondria for Therapy and Diagnosis in Acute Central Nervous System Injury. JAMA Neurol. 2018, 75, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Liu, X.; Wang, B.; Wang, Z.; Liu, Y.; Di, C.; Si, J.; Li, H.; Wu, Q.; Xu, D.; et al. Endocytosis-mediated mitochondrial transplantation: Transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 2019, 9, 3595–3607. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Moon, S.H.; Lee, Y.; Kim, G.J.; Chung, H.M.; Choi, Y.S. Alternative xeno-free biomaterials derived from human umbilical cord for the self-renewal ex-vivo expansion of mesenchymal stem cells. Stem Cells Dev. 2013, 22, 3025–3038. [Google Scholar] [CrossRef]
- Kim, M.J.; Hwang, J.W.; Yun, C.K.; Lee, Y.; Choi, Y.S. Delivery of exogenous mitochondria via centrifugation enhances cellular metabolic function. Sci. Rep. 2018, 8, 3330. [Google Scholar] [CrossRef] [PubMed]
- Dex, S.; Lin, D.; Shukunami, C.; Docheva, D. Tenogenic modulating insider factor: Systematic assessment on the functions of tenomodulin gene. Gene 2016, 587, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuzaki, M.; Guyton, G.; Garrett, W.; Archambault, J.M.; Herzog, W.; Almekinders, L.; Bynum, D.; Yang, X.; Banes, A.J. IL-1 beta induces COX2, MMP-1, -3 and -13, ADAMTS-4, IL-1 beta and IL-6 in human tendon cells. J. Orthop. Res. 2003, 21, 256–264. [Google Scholar] [CrossRef]
- Del Buono, A.; Oliva, F.; Osti, L.; Maffulli, N. Metalloproteases and tendinopathy. Muscles Ligaments Tendons J. 2013, 3, 51–57. [Google Scholar] [CrossRef]
- Stockert, J.C.; Horobin, R.W.; Colombo, L.L.; Blazquez-Castro, A. Tetrazolium salts and formazan products in Cell Biology: Viability assessment, fluorescence imaging, and labeling perspectives. Acta Histochem. 2018, 120, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Mali, V.R.; Pan, G.; Deshpande, M.; Thandavarayan, R.A.; Xu, J.; Yang, X.P.; Palaniyandi, S.S. Cardiac Mitochondrial Respiratory Dysfunction and Tissue Damage in Chronic Hyperglycemia Correlate with Reduced Aldehyde Dehydrogenase-2 Activity. PLoS ONE 2016, 11, e0163158. [Google Scholar] [CrossRef] [PubMed]
- Sadaba, M.C.; Martin-Estal, I.; Puche, J.E.; Castilla-Cortazar, I. Insulin-like growth factor 1 (IGF-1) therapy: Mitochondrial dysfunction and diseases. Biochim. Biophys. Acta 2016, 1862, 1267–1278. [Google Scholar] [CrossRef]
- Hast, M.W.; Zuskov, A.; Soslowsky, L.J. The role of animal models in tendon research. Bone Jt. Res. 2014, 3, 193–202. [Google Scholar] [CrossRef]
- Chisari, E.; Rehak, L.; Khan, W.S.; Maffulli, N. Tendon healing in presence of chronic low-level inflammation: A systematic review. Br. Med. Bull. 2019, 132, 97–116. [Google Scholar] [CrossRef]
- Robertson, C.M.; Chen, C.T.; Shindle, M.K.; Cordasco, F.A.; Rodeo, S.A.; Warren, R.F. Failed healing of rotator cuff repair correlates with altered collagenase and gelatinase in supraspinatus and subscapularis tendons. Am. J. Sports Med. 2012, 40, 1993–2001. [Google Scholar] [CrossRef]
- Tempfer, H.; Traweger, A. Tendon Vasculature in Health and Disease. Front. Physiol. 2015, 6, 330. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-Garcia, C.; Valcarcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef]
- Meyer, A.; Laverny, G.; Bernardi, L.; Charles, A.L.; Alsaleh, G.; Pottecher, J.; Sibilia, J.; Geny, B. Mitochondria: An Organelle of Bacterial Origin Controlling Inflammation. Front. Immunol. 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Vaamonde-Garcia, C.; Riveiro-Naveira, R.R.; Valcarcel-Ares, M.N.; Hermida-Carballo, L.; Blanco, F.J.; Lopez-Armada, M.J. Mitochondrial dysfunction increases inflammatory responsiveness to cytokines in normal human chondrocytes. Arthritis Rheum. 2012, 64, 2927–2936. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Andreu, A.L.; Musumeci, O.; Bonilla, E. Diseases of oxidative phosphorylation due to mtDNA mutations. Semin. Neurol. 2001, 21, 251–260. [Google Scholar] [CrossRef]
- Gallo, J.; Raska, M.; Kriegova, E.; Goodman, S.B. Inflammation and its resolution and the musculoskeletal system. J. Orthop. Transl. 2017, 10, 52–67. [Google Scholar] [CrossRef]
- Alberton, P.; Dex, S.; Popov, C.; Shukunami, C.; Schieker, M.; Docheva, D. Loss of tenomodulin results in reduced self-renewal and augmented senescence of tendon stem/progenitor cells. Stem Cells Dev. 2015, 24, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Docheva, D.; Hunziker, E.B.; Fassler, R.; Brandau, O. Tenomodulin is necessary for tenocyte proliferation and tendon maturation. Mol. Cell. Biol. 2005, 25, 699–705. [Google Scholar] [CrossRef] [Green Version]
- Riley, G. Chronic tendon pathology: Molecular basis and therapeutic implications. Expert Rev. Mol. Med. 2005, 7, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Olejarz, W.; Lacheta, D.; Kubiak-Tomaszewska, G. Matrix Metalloproteinases as Biomarkers of Atherosclerotic Plaque Instability. Int. J. Mol. Sci. 2020, 21, 3946. [Google Scholar] [CrossRef]
- John, T.; Lodka, D.; Kohl, B.; Ertel, W.; Jammrath, J.; Conrad, C.; Stoll, C.; Busch, C.; Schulze-Tanzil, G. Effect of pro-inflammatory and immunoregulatory cytokines on human tenocytes. J. Orthop. Res. 2010, 28, 1071–1077. [Google Scholar] [CrossRef]
- Mehr, D.; Pardubsky, P.D.; Martin, J.A.; Buckwalter, J.A. Tenascin-C in tendon regions subjected to compression. J. Orthop. Res. 2000, 18, 537–545. [Google Scholar] [CrossRef]
- Gottlieb, E.; Armour, S.M.; Harris, M.H.; Thompson, C.B. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003, 10, 709–717. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Villanueva-Paz, M.; de la Cruz-Ojeda, P.; de la Mata, M.; Cotan, D.; Oropesa-Avila, M.; de Lavera, I.; Alvarez-Cordoba, M.; Luzon-Hidalgo, R.; Sanchez-Alcazar, J.A. Mitochondrial Dynamics in Mitochondrial Diseases. Diseases 2016, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [Green Version]
- Jezek, J.; Cooper, K.F.; Strich, R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants 2018, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gan, X.; He, Y.; Zhu, Z.; Zhu, J.; Yu, H. Drp1-dependent mitochondrial fission mediates osteogenic dysfunction in inflammation through elevated production of reactive oxygen species. PLoS ONE 2017, 12, e0175262. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Han, D.; Zheng, X.; Wang, X.; Jin, T.; Cui, L.; Chen, Z. Mesenchymal Stem/Stromal Cell-Mediated Mitochondrial Transfer and the Therapeutic Potential in Treatment of Neurological Diseases. Stem Cells Int. 2020, 2020, 8838046. [Google Scholar] [CrossRef]
- Plachel, F.; Heuberer, P.; Gehwolf, R.; Frank, J.; Tempfer, H.; Lehner, C.; Weissenbacher, N.; Wagner, A.; Weigl, M.; Moroder, P.; et al. MicroRNA Profiling Reveals Distinct Signatures in Degenerative Rotator Cuff Pathologies. J. Orthop. Res. 2020, 38, 202–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterbourn, C.C. The challenges of using fluorescent probes to detect and quantify specific reactive oxygen species in living cells. Biochim. Biophys. Acta 2014, 1840, 730–738. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.M.; Hwang, J.W.; Kim, M.J.; Jung, S.Y.; Kim, K.-S.; Ahn, E.H.; Min, K.; Choi, Y.-S. Mitochondrial Transplantation Modulates Inflammation and Apoptosis, Alleviating Tendinopathy Both In Vivo and In Vitro. Antioxidants 2021, 10, 696. https://doi.org/10.3390/antiox10050696
Lee JM, Hwang JW, Kim MJ, Jung SY, Kim K-S, Ahn EH, Min K, Choi Y-S. Mitochondrial Transplantation Modulates Inflammation and Apoptosis, Alleviating Tendinopathy Both In Vivo and In Vitro. Antioxidants. 2021; 10(5):696. https://doi.org/10.3390/antiox10050696
Chicago/Turabian StyleLee, Ji Min, Jung Wook Hwang, Mi Jin Kim, Sang Youn Jung, Kyung-Soo Kim, Eun Hee Ahn, Kyunghoon Min, and Yong-Soo Choi. 2021. "Mitochondrial Transplantation Modulates Inflammation and Apoptosis, Alleviating Tendinopathy Both In Vivo and In Vitro" Antioxidants 10, no. 5: 696. https://doi.org/10.3390/antiox10050696